This second article describing key pharmacological principles focuses on pharmacodynamics, the physiological effects of drugs on the body. It follows on from the first article, which discussed important pharmacokinetic principles, particularly in relation to anaesthetic and analgesic drugs. It is important to have knowledge of both pharmacokinetics and pharmacodynamics in order to fully understand the effects of individual drugs on the body.
Drug receptors
Generally, in order for a drug to exert an effect on the body, the drug must bind to a receptor. The word ‘receptor’ can be used to describe all target molecules for drug binding, and it is used in this context in this article. Receptors as a group can, however, be divided up into: true receptors, which are usually protein molecules on cell membranes; enzymes, which can be located intra or extracellulary; ion channels in cell membranes; and transport proteins, which are located in cell membranes.
Once the drug has bound to the receptor there is a transduction pathway that precedes drug effect, the time lag between drug binding and drug effect depends on the nature of the transduction pathway.
Transduction pathways
Three different types of transduction mechanism that are common throughout the body are listed below.
- The receptor is linked to an ion channel, for example the acetylcholine nicotinic receptor present at the neuromuscular junction. Binding of acetylcholine to the receptor at two sites causes a structural change in the receptor and the ion channel opens. There is a minimal time lag between binding of the drug to the receptor and drug effect.
- G-protein linked family of receptors: binding of the drug to the receptor causes a conformational change in the receptor, which triggers interaction with a G protein. The activated G proteins stimulate a cascade of events which eventually cause a downstream change in the cell. For example, some G proteins are coupled to adenylate cyclase which catalyzes the conversion of ATP to cAMP. Alpha and beta adrenoreceptors are examples of receptors for hormones that are linked to G proteins.
- Phospholipase C and IP3 transduction pathway: receptors that activate this pathway are mainly G-protein coupled receptors, however, instead of activating the cAMP pathway this sub group of G-protein receptors activates phospholipase C. Phospholipase C cleaves a phospholipid resulting in the production of IP3. IP3 diffuses through the cell to bind to IP3 receptors causing the cytosol concentration of calcium to increase generating a cascade of intracellular changes and activity.
Drug properties
It is possible to define a number of different properties of drugs in relation to binding to the receptor and action within the cell. It is important to have an understanding of these concepts because they relate to the inherent properties of all drugs:
- Agonist — a drug that binds to a receptor and changes the conformational state of the receptor, leading to a biological response. Morphine is an example of a drug that is an agonist at the μ opioid receptor
- Antagonist — a drug that inhibits the action of an agonist. Naloxone is an example of a drug that is an antagonist at the μ opioid receptor
- Efficacy — the characterization and quantification of the ability of agonists to induce a response. For example a full agonist will induce the maximum response that the tissue is capable of following activation of that receptor type (e.g. effect of morphine at the μ opioid receptor). A partial agonist will not induce the maximal response, but a response that is somewhere between no response (elicited by an antagonist) and the maximal response. Buprenorphine is an example of a partial agonist that is less efficacious than morphine and therefore provides less analgesia compared with morphine
- Potency — it is important to understand the difference between drug potency and drug efficacy as sometimes they are used wrongly, and/or interchangeably. Potency is used to quantify the amount of drug required to elicit a given level of response. The lower the concentration required, the greater the potency, however, potency does not relate to what the maximal response is (efficacy). For example, buprenorphine is more potent than morphine but less efficacious as an analgesic agent.
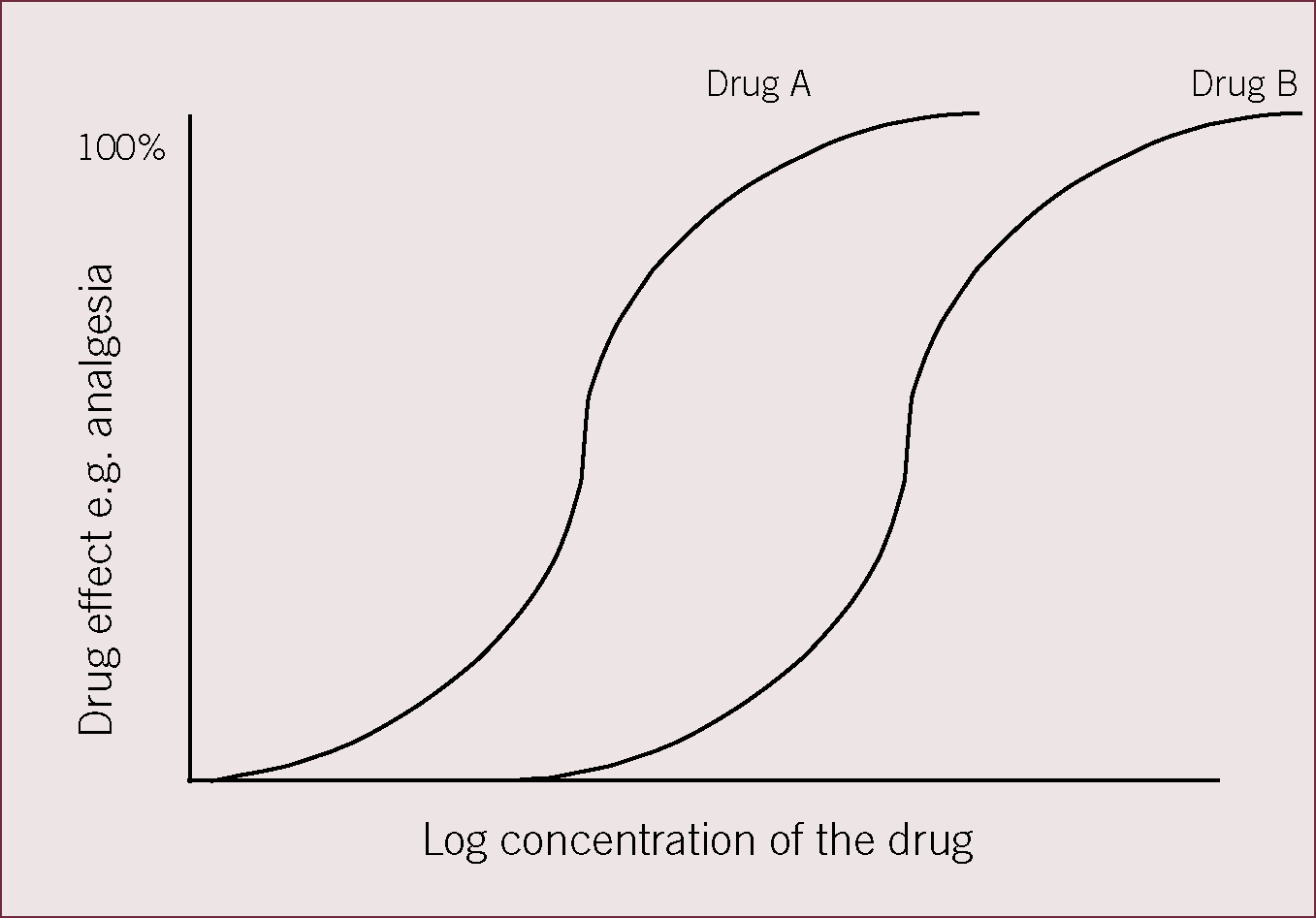
Figure 1 shows drugs A and B are equally efficacious, i.e. they both produce 100% analgesia, but drug B is less potent that drug A because a higher concentration of drug B is required to produce 100% drug efficacy.
Delays in drug effect following drug administration
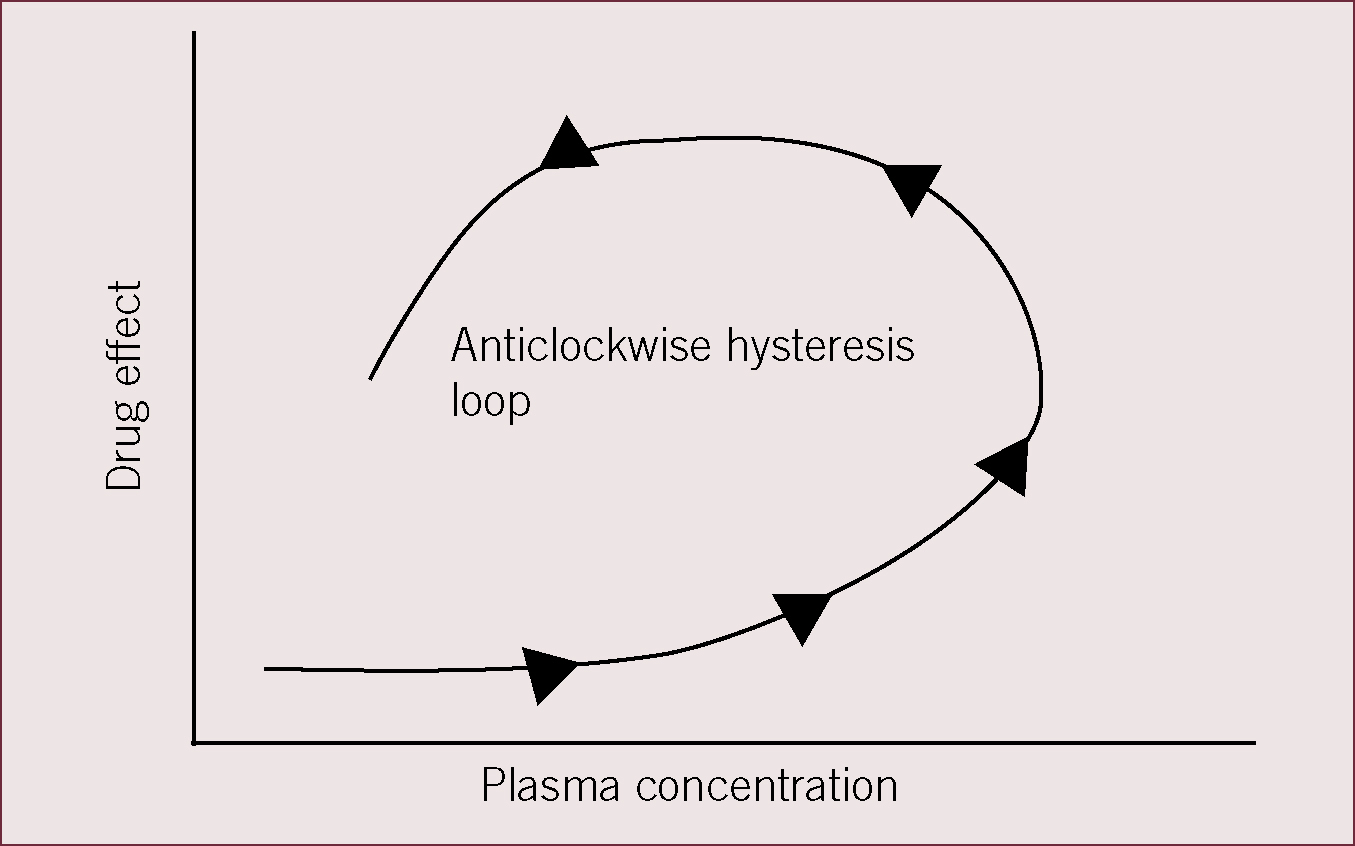
It is not uncommon to observe a delay between achieving a given drug concentration in the plasma and achieving the expected drug response. This may be due to the transduction mechanism and the time taken for the drug to exert an effect. For example, for drugs that exert an effect by stimulating DNA transcription to generate new proteins, the delay in drug effect can be measured in hours. However, an alternative common reason for delays in drug effect can be the fact that the site of drug action is not the plasma, but a different biophase. Buprenorphine must penetrate the blood–brain barrier to enter the central nervous system in order to bind to μ opioid receptors and exert an analgesic effect, therefore, the relationship between plasma concentration and drug effect is represented by a hysteresis loop.
Figure 2 shows a hysteresis loop of drug effect (on the y axis) versus plasma concentration (x axis). At any given plasma concentration of the drug, drug effect may be lower or higher depending on the time after drug administration and whether the drug has entered the biophase to exert its effects or not.
Drug metabolism and elimination
Metabolism
In order to allow drugs to be excreted from the body, which commonly occurs via excretion in the urine, the drug must be a polar or charged molecule. If the molecule is not charged it will usually be reabsorbed in the tubules of the kidney, limiting the capacity for excretion. Therefore the majority of the drugs are metabolized by the liver before they are excreted in order to change them from an uncharged hydrophobic (lacking affinity for water) molecule to a hydrophilic (attracted to water and will usually dissolve in water) charged molecule.
Drug metabolism is commonly divided into phase 1 and phase 2 reactions:
Phase 1: conversion of a drug molecule to a more hydrophilic metabolite
Phase 2: conjugation of the metabolite with a large polar molecule before excretion.
Phase 1 reactions
These usually occur in the liver and are carried out by enzymes of the cytochrome P450 enzyme family. The process involves generation of an oxygen molecule that is able to interact with the organic substrate in various ways, with the result that a larger polar molecule is formed. This molecule may be excreted directly in the urine via the kidneys or may undergo further conjugation.
Phase 2 reactions
The most common conjugation reactions are: glucoronidation; acetylation; sulfation; glutathione.
These reactions increase the polarity of the drug substrate and therefore facilitate renal elimination. It is important to remember that cats are deficient in the metabolic pathway leading to glucoronidation, and therefore drugs that require glucoronidation may be more slowly metabolized in cats. An example of a drug metabolized by glucoronidation is propofol, which is metabolized more slowly in cats than dogs resulting in a longer recovery time following administration to cats compared with dogs.
Drugs that have undergone conjugation may also be excreted in bile and therefore enter the gastrointestinal tract. This opens the possibility of enterohepatic recirculation of drugs, which results in a later peak increase in plasma concentration of the drug as the plasma drug concentration is otherwise falling. Enterohepatic recirculation occurs when bacteria in the small intestine deconjugate excreted drugs, allowing them to be reabsorbed into the systemic circulation via the liver. This prolongs the length of time that the drug is in the body.
Elimination
Most drugs are eliminated from the body by the liver or kidneys via the bile or urine respectively following drug metabolism. However, it should be remembered that some drugs may also be excreted through the lungs, skin or breast milk. Drugs that cannot be rendered water soluble by the process of drug metabolism are generally excreted in the bile which contains sufficient cholesterol to dissolve fat soluble products of drug breakdown. The kidneys are responsible for the elimination of most water soluble drugs via filtration from the plasma by the glomeruli and excretion into the urine. The rate of drug excretion depends on the glomerular filtration rate. Water soluble drugs may also be actively secreted by the kidney by excretion into the renal tubules.
Clearance and half life are two pharmacokinetic parameters that are used to describe the exponential fall in plasma concentration of a drug against time and can both be used to design drug dosing strategies required to maintain a ‘set’ plasma concentration of a drug.
Half life (t½): This is the time taken for the concentration of the drug in the bloodstream to fall to half of its original value. Drugs given by continuous intravenous infusion need to have a short half life to avoid drug accumulation resulting from continuous administration. Half life also defines the redosing interval.
Clearance: This is the volume of blood or plasma that is cleared of the drug in unit time and it gives an indication of the ability of the liver and kidney to remove the drug from the plasma or blood. Clearance, half life and volume of distribution (Vd) are related to each other by the following equation:
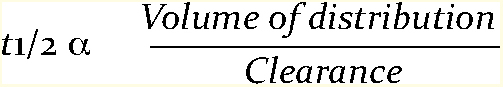
The practical consequences of this relationship are that the half life of a drug will be short if the drug has a small Vd and a high clearance, whereas the half life will be long if the drug is widely distributed throughout the body (large Vd) and clearance by the liver and kidneys is slow.
Relationship between pharmacodynamics and pharmacokinetics
It is important to study the relationship between drug pharmacodynamics and kinetics in order to draw conclusions about drug dosing, dosing interval and plasma concentration of the drug required to produce drug effect. For example, in studies of analgesia, the analgesic effect of a drug cannot be inferred by simply measuring the plasma concentration of the drug in a veterinary species and extrapolating from human pharmacokinetic data (KuKanich and Papich, 2004). It is important that both kinetic and dynamic data are collected in the same study so that drug effect (onset of action, offset of action and peak effect) can be related to change in plasma concentration over time. This allows meaningful and accurate recommendations about drug dose and dosing interval required to produce and maintain analgesia to be made.
Conclusions
Pharmacodynamics and kinetics are subjects that lie at the core of understanding drug action on the body and as such are important to understand, particularly in the field of anaesthesia and analgesia. This series of two articles introduces some of the basic principles of dynamics and kinetics but does not attempt to be a comprehensive review of the subject. It is hoped that it will form a platform for further reading in this area.